Getting Started
This page documents some of the standard operating procedures used by members of the GO Consortium during the process of annotation. Please note that these represent the best, or only ways to carry out annotation, but are simply a guide to how some groups currently annotate. More information on annotation can be found in the GO annotation guide and in the GO annotation conventions; if you have any questions on the guidelines given below, please email the GO helpdesk.
Tell us about your requirements
I represent a small lab working on biological area
In this case, perhaps you have a list of your favourite genes and you wish to annotate them. You have a range of choices depending on what you are trying to achieve.
Please see the range of options below and choose the one that suits you best.
I have a set of ESTs and I would like to attach annotations
If you would like ultimately to send the annotations to the consortium for distribution then it is crucial that your EST clusters should maintain the same identifiers over each round of reclustering. One way to do this is to identify clusters based on one EST that is chosen for each cluster. There may be other good ways that we have not heard of.
Many EST clusters have stable identifiers with version updates (e.g. the UniGene database). These stable identifiers can be used for making GO associations.
Once you have your clusters and stable identifiers follow the IEA directions for making electronic annotation.
You could also run BlastX, or run gene prediction programmes and then BlastP. Running InterPro on the sequences will find the longest open reading frame.
I have a genome sequence
You will already have assembled the genome sequence and made gene calls. Once you have the cds sequences or predicted protein sequences then you can follow the instructions on IEA annotation and/or Literature annotation.
I have a microarray data set
The action you can take depends somewhat on your sequences.
- Are they cDNAs or oligos?
- Do they have identifiers? Which kind?
- How do they relate to the genes? If you know which sequence relates to which characterised gene then it will be easy to transfer annotations over.
- Do the genes have GO annotations? If they do not have full GO annotation from literature then you may like to apply for funding to annotate the genes yourself, or write to your Model Organism Database to ask them to do so.
- Can you get more up to date annotations than those provided with your tool? It may be that you are seeing only the annotations that come from your proprietary microarray software provider. It is a good idea to ask how often they update their annotations and ontology structure as these change from day to day, and there may be many more annotations available than you are seeing.
It is most likely that you will want to use mainly electronic annotations, supplemented with some literature annotation for those sequences that are not yet fully annotated.
I have a peptide sequence
- Do you know what gene is it?
- Can you map it to a UniProtKB or MOD identifier?
- Does this identifier have GO annotation?
If it doesn't, you can request that it be annotated (it helps if you provide literature associated with this gene product). If you cannot map it to a UniProtKB or MOD identifier, then you can make your own GO annotation by any of the electronic or ISS methods illustrated below.
Electronic annotation
Electronic annotation is very quick and produces large amounts of less detailed annotation very quickly. Electronic annotations are rarely wrong, but tend to be less detailed. For example, electronic annotation is likely to tell you which of your genes are transcription factors but unlikely to tell you in great detail what process the gene controls. You may like to use this method if you have a new genome sequence to annotate, or a microarray with many thousands of sequences.
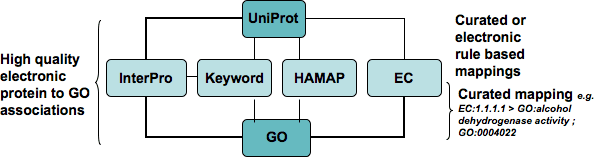
This diagram illustrates some of the main ways of making electronic annotation. It should be read from the top down. The diagram shows sequences from UniProt having electronic GO annotation assigned by several computational methods. All of these methods involve use of mapping files. For more information on mappings see the information on mappings of GO to other classification systems.
InterPro Mapping
In the case of the Interpro mapping it is possible to assign electronic GO annotation to your sequences based on InterPro domains and a number of other criteria. For example if your sequence has a DNA binding domain then it makes sense to electronically annotate it to the DNA binding function term. For more information on InterPro mapping please see the information on InterProScan.
UniProt Keyword Mapping
This part of the diagram illustrates how sequences already categorised using the UniProt keyword mapping can have GO annotation automatically applied by transferring via the keyword mapping file.
HAMAP
HAMAP is a system that categorizes sequences based on family or subfamily characteristics and is applied to bacterial, archaeal and plastid-encoded proteins. GO annotation can be automatically applied to such sequences using the mapping file between HAMAP and GO.
Enzyme Commission
The Enzyme Commission database categories enzymes by the reactions they catalyse. If your sequences are already categories by EC then you can transfer GO annotations using the mapping file of EC to GO categories.
Other mappings
These are just a few examples of mapping files that can be used to transfer annotations to your sequence objects. Many other mappings are available, and if there is not a mapping file between GO and your current annotation system, we can assist you in making one.
BLAST
You can also make electronic annotations by BLASTing your sequence against manually annotated sequences and transferring the GO annotations across to your sequence. The threshold of similarity in this process is up to you, and depends on your requirements.
No similar sequences manually annotated?
If your sequence is similar to other sequences that have been well characterised but not yet annotated from the literature, then one option is to carry out the literature annotation yourself and then transfer by electronic methods.
Literature Annotation
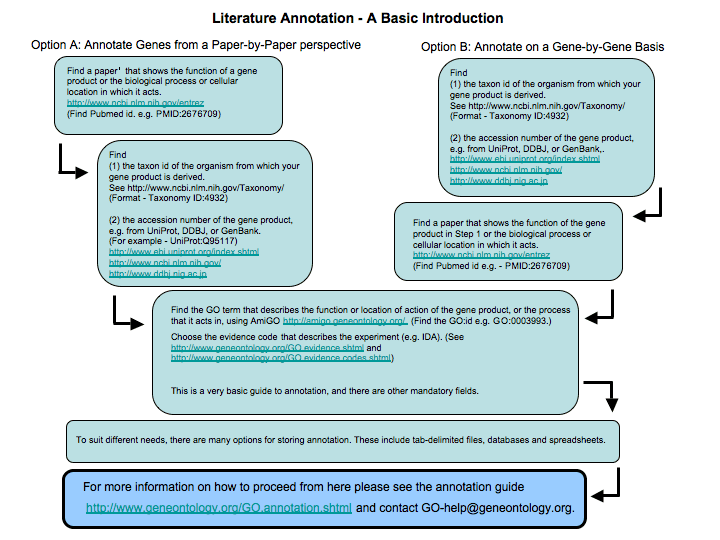
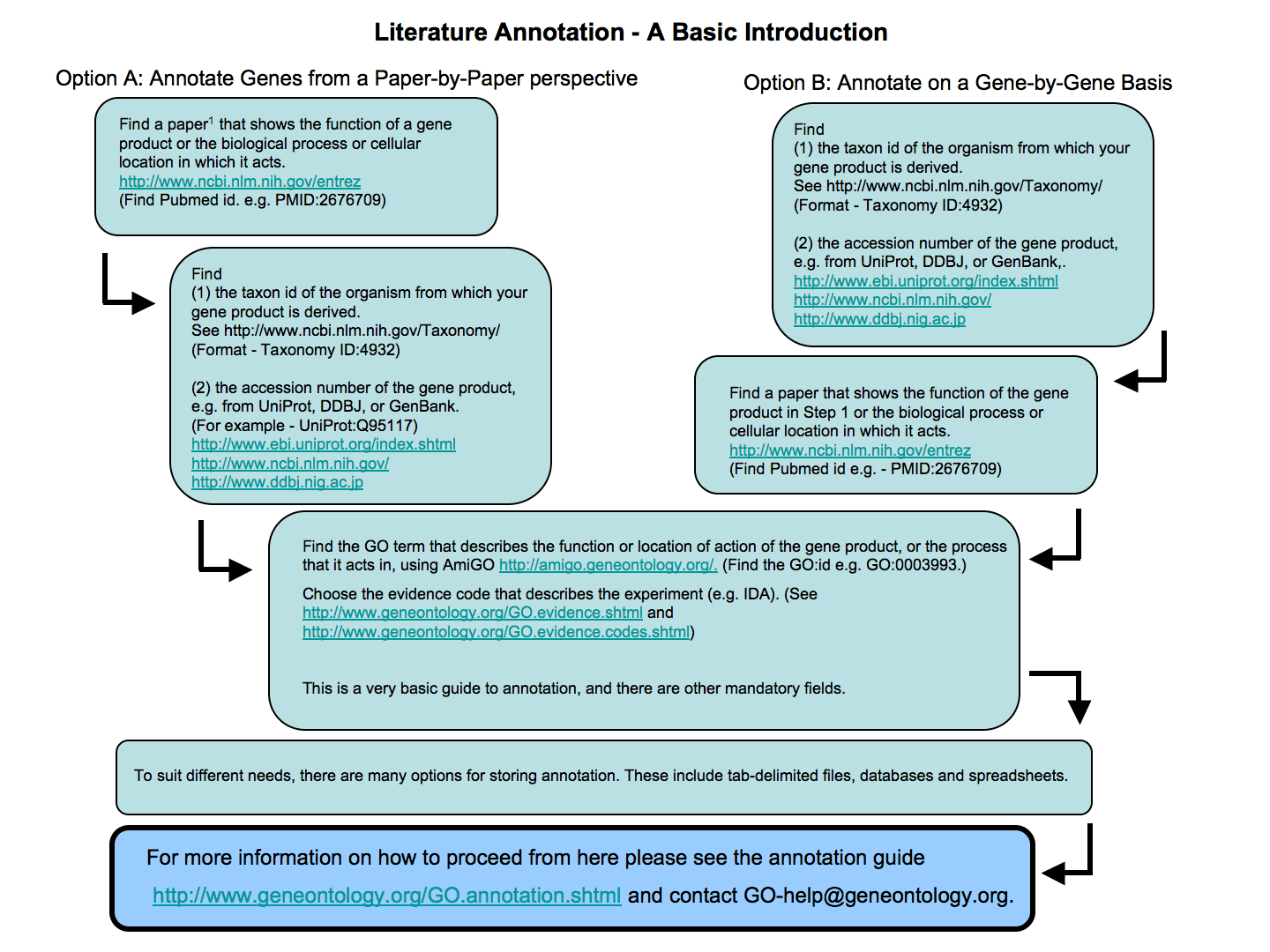
Literature annotation involves capturing published information about the exact function of a gene product as a GO annotations. To do this you must read the publications about the gene and write down all the information. This annotation is time-consuming but produces very high quality, species-specific annotation, and brings the information about the gene product into a format in which it can be used in high-throughput experiments. This is an extremely worthwhile process in the long term. It may be best carried out by people who know the function of the gene product, and the associated biology, in great detail; for example experimental scientists who are familiar with the published literature. If you are doing this, then you may like to write and suggest modifications to the ontology structure as well.
Below is a schematic diagram giving an introduction to the steps involved in literature-based GO annotation. If you are interested in carrying out literature-based annotation you can receive full training in the process by attending a GO annotation camp or by working with an individual GO Consortium annotation mentor.
{kind=link}
Sequence-based annotation
General principles for sequence IDs
- You must have stable identifiers for your objects.
- You must provide information on what the object is, e.g. a protein, nucleotide, EST, etc.. It doesn't matter if a nucleotide sequence is a gene, a genome, or an EST as long as it can be identified as such.
- If a sequence identifier has become obsolete, there must be a mechanism in place for tracking down the replacement.
- Your database must have an internal rule that object identifiers are never reused.
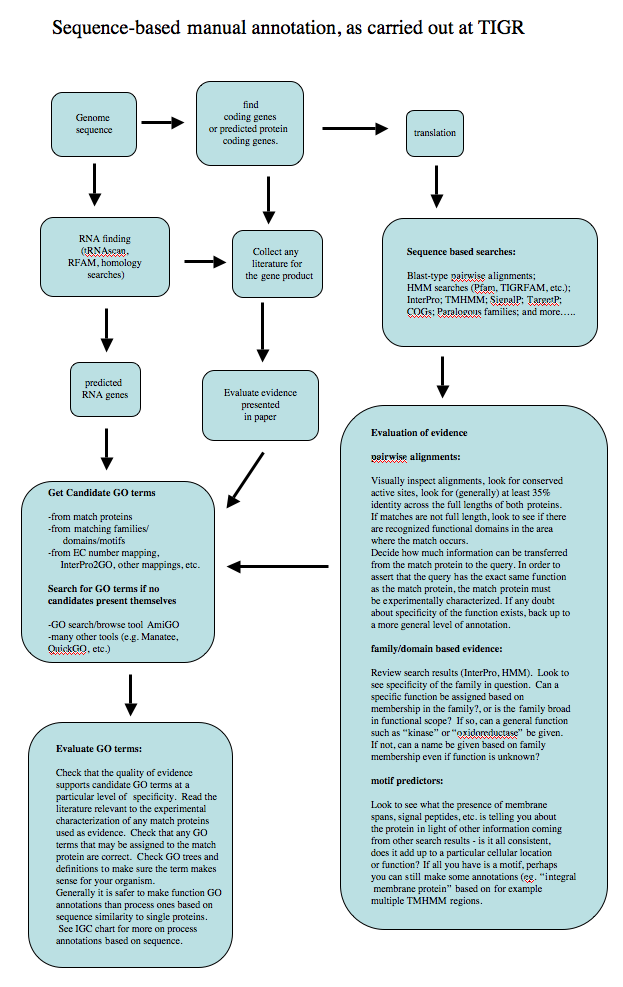
Annotation workflow
The following diagram shows the standard operating procedure for sequence-based (ISS evidence code) annotation used in the past at The Institute for Genomic Research (now JCVI).